ABSTRACT
Introduction: Furosemide is a widely used loop diuretic for managing conditions such as oedema and hypertension. In paediatric patients, especially in the absence of commercially available liquid formulations, extemporaneous oral suspensions are often prepared. This study aimed to evaluate the stability of furosemide oral suspensions formulated at two different concentrations, made from commercial tablets using the X-Temp® Oral Suspension System. Methods: Furosemide suspensions were prepared at concentrations of 2 mg/mL and 10 mg/mL using the X-Temp® system, then packaged in amber HDPE bottles. The suspensions were stored under refrigeration (5 ± 3°C) or at room temperature (30 ± 2°C) for a duration of up to 90 days. A comprehensive evaluation protocol, including visual inspections, pH measurements, and HPLC assays, provided a multi-faceted assessment of the samples’ stability over the 90-day period. The inclusion of microbiological testing at each interval further reinforced the safety profile of the product. A validated HPLC method was used to determine furosemide content in the oral suspension, ensuring the accuracy, precision, and reliability of the analytical data provided by the system. Results: The results showed no statistically significant changes in the assay of furosemide suspension at concentrations of 2 mg/mL and 10 mg/mL over 90 days (n = 3, p > 0.05). In addition, the stability studies revealed that furosemide in X-Temp® suspension was physically and microbiologically stable throughout the study period. HPLC analysis results shows that all the samples maintained drug concentrations within the specified range. Therefore, it can be concluded that furosemide tablets can be compounded extemporaneously using the X-Temp® Oral Suspension System at concentrations between 2 mg/mL and 10 mg/mL. The resulting suspensions remain stable for up to 90 days when stored in amber HDPE bottles at either 5 ± 3°C or 30 ± 2°C. Conclusion: This study provides important insights into the stability of extemporaneous furosemide oral suspensions compounded using the X-Temp® Oral Suspension System. The findings offer valuable guidelines for the preparation, storage, and use of furosemide in X-Temp® suspensions in paediatric care, highlighting the need for careful consideration of storage conditions to ensure therapeutic efficacy.
INTRODUCTION
Furosemide, a potent loop diuretic, is a cornerstone in the treatment of conditions such as oedema associated with heart failure, liver cirrhosis, and renal diseases, as well as in managing hypertension [1]. Despite the availability of furosemide in tablet and intravenous forms, there is limited access to liquid formulations suitable for paediatric patients, who often require precise dosing and may have difficulty swallowing solid dosage forms [1]. This gap necessitates the preparation of extemporaneous oral liquid formulations, particularly in paediatric care.
The pharmacokinetics of furosemide in children differ significantly from those in adults due to the immature organ systems in younger patients [2][3]. These differences underscore the importance of stable and consistent formulations to ensure effective therapeutic outcomes. Given the lack of a commercially available liquid formulation, extemporaneous preparation is often the only viable option for paediatric patients. However, the sensitivity of furosemide towards factors such as pH, light, and temperature [4][5] raises concerns about the stability of the preparation, which could potentially lead to degradation and reduced efficacy.
The X-Temp® Oral Suspension System, a ready-to-use suspending vehicle, offers a practical and efficient option for compounding pharmacists, providing a safe and time-saving alternative. To explore its potential, this study was conducted to evaluate the stability of furosemide oral suspension at two different concentrations, prepared from commercial tablets using the X-Temp® Oral Suspension System. This system is specifically designed to facilitate the preparation of stable and uniform oral suspensions. By systematically evaluating the physicochemical and microbiological properties of these formulations under various storage conditions, this research aims to provide evidence-based guidelines for the preparation, storage, and administration of furosemide in X-Temp® suspensions, with a particular focus on paediatric applications.
METHOD
Oral suspensions were prepared from commercially available tablets, each containing 40 mg of furosemide (Pharmaniaga Frusemide Tablet 40 mg, Malaysia). The X-Temp® Oral Suspension System, marketed by Pharm-D Sdn. Bhd. (Malaysia), was the commercial suspending system utilized in this study. This system is commonly employed in hospitals to support the extemporaneous preparation of oral liquids and aqueous formulations with insoluble components.
Preparation of furosemide in X-Temp® suspension
In this study, two concentrations of furosemide suspension were prepared: 2 mg/mL and 10 mg/mL. For each concentration, forty bottles containing 100 mL of the suspension (totalling 4000 mL) were prepared. Before levigating, the required amount of X-Temp® Oral Suspension System was measured, and the furosemide tablets were ground into a powder using a mortar and pestle. A small portion of X-Temp® Oral Suspension System was initially mixed with the powder to create a smooth paste. The remaining X-Temp® Oral Suspension System was then gradually incorporated into the paste while stirring continuously until a uniform liquid was obtained. Subsequently, the mixture was transferred to a graduated container, and the mortar and pestle were rinsed thoroughly using the X-Temp® Oral Suspension System. The final volume of 4000 mL was achieved by adding more X-Temp® Oral Suspension System. To simulate actual dispensing conditions, the prepared bulk suspension was transferred into 40 amber high-density polyethylene (HDPE) bottles, each containing 100 mL, and sealed with white polypropylene (PP) screw caps.
Stability Evaluation
In the stability study, two concentrations of furosemide in X-Temp® suspension, 2 mg/mL and 10 mg/mL, were prepared and stored under two different conditions to simulate the dispensing environments. The first group was kept at room temperature, specifically 30 ± 2°C with a relative humidity of 75 ± 5%, while the second group was stored under refrigeration at 5 ± 3°C. Each group consisted of 18 bottles, and the samples were maintained for 90 days. To evaluate the stability of the samples, aliquots from both the 2 mg/mL and 10 mg/mL concentrations were collected at various time points: Day 0, Day 15, Day 30, Day 45, Day 60, Day 75, and Day 90.
- Physical Stability
The assessment of physical stability through visual inspection was the first step in ensuring the quality of oral suspensions. It involved observing the suspension for any changes in colour, flavour, and clarity, which were indicative of its stability. A stable preparation was defined as one in which these physical characteristics did not change significantly over time. Additionally, the measurement of specific gravity was an important physical test that compared the weight of the sample to that of water [6], providing insight into the concentration and consistency of the suspended drug. This test was conducted at a controlled room temperature to ensure its accuracy and reliability. The specific gravity values for concentrations of 2 mg/mL and 10 mg/mL were expected to fall within the range of 1.01 to 1.07 g/mL, as per the established in-house specifications. This range was critical for confirming that the oral suspension maintained the appropriate density and composition throughout the study period.
- Chemical stability
The concentration of furosemide in the samples is a critical parameter and the specification limit was set to be within 90.0% to 110.0% of the labelled amount to ensure efficacy and safety [7]. The analysis was performed using High-Performance Liquid Chromatography (HPLC), and the retention time (RT) of furosemide in both the reference standard solution and the sample solutions was compared to confirm the identity of the compound. Concurrently, the peak area of furosemide, as the compound of interest, was calculated to provide a quantitative measure of its concentration. This dual assessment of RT and peak area underpinned the capability of HPLC in ensuring the quality of pharmaceutical products.
The concentration of furosemide in the oral suspension was determined using the formula below:

The limit test for 4-chloro-5-sulfamoylanthranilic acid, also known as furosemide related compound B, was a quality control measure in the detection of degradation product for this study. The calculation of the percentage of 4-chloro-5-sulfamoylanthranilic acid in the oral suspension was performed based on the formula shown below:
Percentage of 4-chloro-5-sulfamoylanthranilic acid (%) (furosemide related compound B) in limit test:
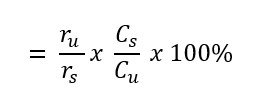
Where
| ru | = | peak response of furosemide related compound B in the sample solution |
| rs | = | peak response of furosemide related compound B in the standard solution |
| Cs | = | concentration of USP Furosemide Related Compound B RS in the standard solution (mg/mL) |
| Cu | = | nominal concentration of furosemide in the sample solution (mg/mL) |
The pH values of the samples were measured at each time point by tilized a digital pH meter (Metrohm Model 913, Metrohm, Switzerland). The study outcomes were documented appropriately.
Analytical Method and Equipment
The quantification of furosemide was performed using HPLC equipped with a UV/Vis detector (Agilent, United States). The furosemide reference standard used was a working standard tilizedize against a primary reference standard obtained from the European Directorate for the Quality of Medicines & HealthCare (EDQM).
Table I. HPLC Operating Conditions.
| Description | Parameters |
|---|---|
| Type of Column | Zorbax Exclipse® XDB-C8 |
| Column size | 4.6 mm × 150 mm, 5 µm |
| Detection | UV 238 nm |
| Flow rate | 1.0 |
| Injection volume | 10 µL |
| Mobile phase | 0.021 M Potassium Dihydrogen Orthophosphate (KH2PO4) pH 7.00 with Cetrimide and Propan-2-ol (62:38 v/v) |
Using the British Pharmacopoeia as a guide, a HPLC assay method was established based on the internal HPLC procedure [6]. The compound of interest was eluted with a mixture of 0.021 M potassium dihydrogen orthophosphate (KH2PO4) at pH 7.00, combined with cetrimide and propan-2-ol (62: 38, v/v) as the mobile phase. The diluent used in the preparation of reference standard and test sample was a combination of 40% propan-2-ol and 60% 0.021 M potassium dihydrogen orthophosphate (KH2PO4) at pH 7.00 with cetrimide. The furosemide peak in the sample chromatograph was compared to the furosemide peak of the reference standard chromatograph. The operating conditions applied in the reversed-phase HPLC assay analysis were tilizedi in Table I.
The methods employed In this study were previously validated in a short-term stability investigation of extemporaneously prepared furosemide oral suspension using the same formulation. The validation process for the analytical methods encompassed specificity, linearity, accuracy, precision, and system suitability.
Identification and assay of furosemide using HPLC
- Preparation of reference standard
Approximately 100 mg of furosemide reference standard was dissolved in roughly 70 mL of diluent in a 100 mL amber volumetric flask and subjected to sonication for 5 minutes. The solution was brought to a final volume of 100 mL to yield the standard stock solution. A 10 mL portion of the standard stock solution was then diluted to 50 mL, and 5 mL of the diluted solution was diluted again to 50 mL using diluent. Prior to injection into the HPLC system, the solution was filtered through a 0.2 µm syringe filter into the an amber autosampler vial.
- Preparation of Test Sample
To prepare a stock solution with a concentration of 2 mg/mL, a weighed quantity of the furosemide suspension containing 10 mg of furosemide was mixed with 70 mL of diluent. The solution was capped and subjected to sonication in a water bath for 5 minutes before being diluted to 100 mL with the diluent. A total of 10 mL of the solution was then diluted to 50 mL using the diluent and filtered through a 0.2 µm syringe filter into an amber autosampler vial for chromatographic analysis.
To prepare a stock solution with a concentration of 10 mg/mL, a weighed quantity of the furosemide oral suspension containing 50 mg of furosemide was mixed with 70 mL of diluent. The solution was capped and subjected to sonication in a water bath for 5 minutes before being diluted to 100 mL with the diluent. The sample solution was diluted to 100 mL with the diluent. From this solution, 10 mL was further diluted to 50 mL with diluent, another 10 mL from the resulting solution was again diluted to 50 mL with diluent and filtered through a 0.2 µm syringe filter into an amber autosampler vial for chromatographic analysis. The resulting concentration of the standard and sample solutions prepared was 0.02 mg/mL.
Limit Test of 4-chloro-5-sulfamoylanthranilic acid (furosemide related compound B)
A series of test solutions was prepared for the quantification of 4-chloro-5-sulfamylanthranilic acid in the study samples. A system suitability solution was prepared with 0.1 mg/mL of furosemide and furosemide related compound A, and 0.015 mg/mL of furosemide related compound B as standard solution. A sensitivity solution of 0.0015 mg/mL of furosemide related compound B was also prepared for the limit test analysis. The sample test solution at a concentration of 1 mg/mL was prepared for the limit test. Matrix solution and diluent were also included in the limit test analysis for peak comparison. All sample solutions were subjected to filtration through a 0.2 µm syringe filter before being injected into the HPLC system. The limit test of furosemide related compound B was analysed through reversed-phase HPLC. The operating conditions applied in the limit test were tilizedi in Table II.
Table II. HPLC Operating Conditions for Limit Test.
| Description | Parameters |
|---|---|
| Type of Column | Zorbax SB-CN |
| Column size | 4.6 mm × 250 mm, 5 µm |
| Detection | UV 254 nm |
| Flow rate | 1.0 |
| Injection volume | 10 µL |
| Mobile phase | 17.3% Acetonitrile 82.7% Mobile phase mixture [Acetonitrile: Glacial acetic acid: Water (22:1:22)] |
Microbiological stability
To ascertain whether the samples conformed to the microbiological attributes of non-sterile pharmaceutical, microbiological evaluation was performed on the samples in accordance with BP2024. According to BP2024 [6], the microbiological test parameters were set as Total Aerobic Microbial Count (TAMC) below 2 × 102 CFU/g, Total Combined Yeasts and Moulds Count (TYMC) below 2 × 10 CFU/g, and absence of Escherichia coli (E. coli) in 1 g.
- Microbial Enumeration Tests
The spread plate technique was employed to determine the TAMC and TYMC. A 10 g sample was diluted in 90 mL of buffered sodium chloride-peptone solution (pH 7.0). A 1:10 dilution was prepared, and a 1% w/v solution of polysorbate 80, used to inactivate antimicrobial agents, was added to the mixture. Serial tenfold dilutions (10-1, 10-2, etc.) were then prepared using the same diluent to achieve a colony count between 25 and 250. From these dilutions, 0.5 mL of each was transferred onto two Petri dishes containing Soybean-Casein Digest Agar (TSA) and two dishes with Sabouraud Dextrose Agar (SDA). The solution was evenly spread across the surface, and the plates were allowed to dry. Subsequently, the TSA plates were covered, inverted, and incubated at 30 – 35°C for 3 – 5 days to assess total aerobic bacterial and fungi, while the SDA plates were incubated at 20 – 25°C for 5 – 7 days to evaluate yeast and mould. The plates were then inspected for signs of microbial growth. A negative control was prepared concurrently using tilizedi diluent in place of the sample for each medium.
- Tests for Escherichia coli
A 10 g sample of furosemide oral suspension was diluted in 90 mL of buffered sodium chloride-peptone solution (pH 7.0). A 1:10 dilution was prepared, and 1% w/v polysorbate 80, an antimicrobial inactivator, was added to the mixture. To inoculate 100 mL of Soybean-Casein Digest Broth, 10 mL (equivalent to 1 g or 1 mL) of the diluted sample was used. The broth was homogenized and left to incubate at 30 – 35°C for 18 – 24 hours. After the incubation period, the broth was agitated to ensure proper mixing, and 1 mL of the Soybean-Casein Digest Broth was transferred into 100 mL of the MacConkey Broth. This was then incubated at a temperature range of 42 – 44°C for 24 – 48 hours. Following this, the MacConkey Broth was shaken, and a loopful was streaked onto MacConkey Agar plates using a sterile inoculating loop. The plates were incubated inverted at 30 – 35°C for 18 – 72 hours. Colony growth was examined for the presence of red nonmucoid colonies on the MacConkey Agar. The isolates were tilizedized using Gram staining and biochemical tests, with further confirmation using the API 20E (bioMérieux) identification system.
Statistical Analysis
The assay of furosemide oral suspension was performed in triplicate, and the results were presented as mean ± SD (Standard Deviation) and analysed using one-way ANOVA. GraphPad Prism X and Microsoft Excel 2019 were utilised to conduct statistical analysis.
RESULTS AND DISCUSSION
Furosemide oral suspensions at the concentrations of 2 mg/mL and 10 mg/mL were compounded from commercially available furosemide tablets using the X-Temp® Oral Suspension System, a commercially produced suspending agent. The extemporaneous suspension was prepared through a straightforward and reproducible process, following current good manufacturing practices for pharmaceutical compounding. Validation of the method was performed to ensure that it was congruous with its intended purpose, thereby providing accurate and precise results. Following the ICH Q2 guidelines, a structured approach to validation was employed, which included assessing parameters such as specificity, linearity, range, and precision [8]. The analysis fulfilled all specification criteria during the validation process, further confirming the reliability of the method in the quantification of furosemide in oral suspension formulations. A summary of method validation is tabulated in Table III.
Table III. Results of Method Validation.
| Test Parameter | Acceptance Criteria (6) (8) |
| Specificity | |
| Identification | |
| Comparison with a known reference material | Positive control: furosemide peak at RT 13.0 min |
| Negative control: absence of peak at RT 13.0 min | |
| Assay | |
| Placebo / Matrix analysis | No interfering peak from excipients |
| Placebo Effect NMT 1.5 % | |
| Linearity & Range | r2 ≥ 0.995; 0.016 – 0.064 mg/mL |
| Y-intercept at 100% working concentration ≤ | |
| 2.0 % | |
| Accuracy | % Recovery within 95% to 105 % |
| Precision (Repeatability) | RSD ≤ 2.0 % |
| Precision (Reproducibility) | RSD ≤ 2.0 % |
| Precision (Intermediate Precision / Ruggedness) | RSD ≤ 2.0 % |
| System Suitability | |
| a.System Specificity | RSD ≤ 2.0 % |
| b. Peak identification | k’ ≥ 1.5 USP Tailing Factor < 2 Column efficiency ≥ 2000 |
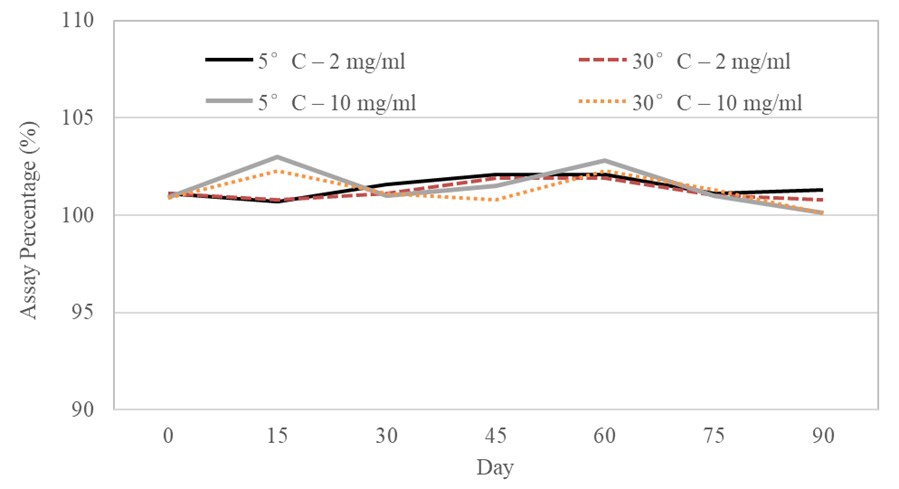
The stability of furosemide in X-Temp® suspension was studied at two different concentrations – 2 mg/mL (low dose) and 10 mg/mL (high dose) – under both room temperature conditions (30 ± 2°C) and refrigerated conditions (5 ± 3°C).
Stability studies revealed that furosemide in X-Temp® suspension, at both low and high doses, remained stable under two storage conditions for at least 90 days. As shown in Figure I, the furosemide level in the samples remained consistent and fell within the established specifications. The results obtained could provide a reference point for the formulation of furosemide in X-Temp® suspension, considering its stability at both low and high dose concentrations.
The physical, chemical, and microbial stability profiles of the furosemide in X-Temp® suspension (2 mg/mL and 10 mg/mL under two different storage conditions) over 90 days are presented in Table IV and Table V. The assay of the 2 mg/mL suspension ranged from 100.7% to 102.1%; for the 10 mg/mL suspension, the result ranged from 100.1% to 103.0%. From the data analysis, all the assay percentages showed no significant difference between the group of samples and remained consistent throughout the 90-day stability study.
- Physical Stability
Throughout the 90-day study period conducted at 2 distinct storage temperatures (5 + 3°C and 30 + 2°C), no significant alterations were identified regarding the colour, flavour, or clarity of suspension. In terms of the specific gravity, there was only negligible change observed during the stability study period.
- Chemical Stability
All samples from both 2 mg/mL and 10 mg/mL were assayed at specific study intervals. The findings reveal that furosemide content in both 2 mg/mL and 10 mg/mL samples were in the range of 100.0% to 103.0% of the labelled amount. Based on the results obtained throughout the 90-day stability study, it could be inferred that furosemide remained stable when suspended in X-Temp® Oral Suspension System.
Table IV: Stability Profile of Furosemide in X-Temp® Suspension 2 mg/mL
| Test | Specifications | Storage condition | Days | ||||||
| 1 | 15 | 30 | 45 | 60 | 75 | 90 | |||
| Visual Appearance | Colour: White to Off- white Clarity: Opaque Flavour: Orange | 5 ± 3°C 30 ± 2°C | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange |
| Physical Properties | pH: 4.0 – 4.4 | 5 ± 3°C 30 ± 2°C | 4.255 4.255 | 4.260 4.266 | 4.256 4.253 | 4.259 4.253 | 4.258 4.244 | 4.254 4.229 | 4.213 4.232 |
| Specific gravity: 1.01 – 1.07 (g/mL) | 5 ± 3°C 30 ± 2°C | 1.0356 1.0356 | 1.0361 1.0377 | 1.0382 1.0348 | 1.0390 1.0380 | 1.0392 1.0371 | 1.0373 1.0368 | 1.0375 1.0375 | |
| Assay* (%) | 90.0 % – 110.0 % | 5 ± 3°C 30 ± 2°C | 101.1 ± 0.79 101.1 ± 0.79 | 100.7 ±1.42 100.8 ± 1.36 | 101.6 ±0.22 101.1 ± 0.37 | 102.1 ± 0.41 101.9 ± 0.37 | 102.1 ± 0.41 101.9 ± 0.08 | 101.1 ± 0.76 101.0 ± 0.62 | 101.3 ± 0.21 100.8 ± 0.57 |
| Related Substances (%) | Furosemide Related Compound B Not more than 1.5% | 5 ± 3°C 30 ± 2°C | 0.01 0.01 | 0.02 0.03 | 0.02 0.02 | 0.01 0.01 | 0.02 0.03 | 0.004 0.02 | 0.008 0.02 |
| Microbial limit | Aerobic microbial: < 200 CFU/g Yeasts & moulds: < 20 CFU/g Escherichia coli: Absent in 1 g | 5 ± 3°C 30 ± 2°C | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms |
Note: *Assay percentage is expressed as mean ± standard deviation of triplicate (n = 3, p < 0.05).
Table V: Stability Profile of Furosemide in X-Temp® Suspension 10 mg/mL
| Test | Specifications | Storage condition | Days | ||||||
| 1 | 15 | 30 | 45 | 60 | 75 | 90 | |||
| Visual Appearance | Colour: White to Off- white Clarity: Opaque Flavour: Orange | 5 ± 3°C 30 ± 2°C | White & Opaque; Orange White & Opaque; Orange | White & Opaque; Orange White & Opaque; Orange | White & Opaque; Orange White & Opaque; Orange | White & Opaque; Orange White & Opaque; Orange | White & Opaque; Orange White & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange | Off-white & Opaque; Orange Off-white & Opaque; Orange |
| Physical Properties | pH: 4.0 – 4.4 | 5 ± 3°C 30 ± 2°C | 4.217 4.217 | 4.239 4.254 | 4.227 4.233 | 4.237 4.239 | 4.229 4.236 | 4.217 4.226 | 4.205 4.188 |
| Specific gravity: 1.01 – 1.07 (g/mL) | 5 ± 3°C 30 ± 2°C | 1.0479 1.0479 | 1.0474 1.0484 | 1.0478 1.0468 | 1.0488 1.0480 | 1.0480 1.0473 | 1.0467 1.0468 | 1.0472 1.0469 | |
| Assay* | 90.0 % – 110.0 % | 5 ± 3°C 30 ± 2°C | 100.9 ± 0.86 100.9 ± 0.86 | 103.0 ± 1.00 102.3 ± 1.80 | 101.0 ± 0.55 101.1 ± 0.55 | 101.5 ± 0.52 100.8 ± 0.26 | 102.8 ± 0.40 102.3 ± 0.58 | 101.0 ± 0.23 101.3 ± 0.39 | 100.1 ± 0.65 100.1 ± 0.13 |
| Related Substances | Furosemide Related Compound B Not more than 1.5% | 5 ± 3°C 30 ± 2°C | 0.10 0.10 | 0.09 0.10 | 0.09 0.10 | 0.08 0.10 | 0.08 0.10 | 0.09 0.12 | 0.08 0.12 |
| Microbial limit | Aerobic microbial: < 200 CFU/g Yeasts & moulds: < 20 CFU/g Escherichia coli: Absent in 1 g | 5 ± 3°C 30 ± 2°C | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms | Conforms Conforms |
Note: *Assay percentage is expressed as mean ± standard deviation of triplicate (n = 3, p < 0.05).
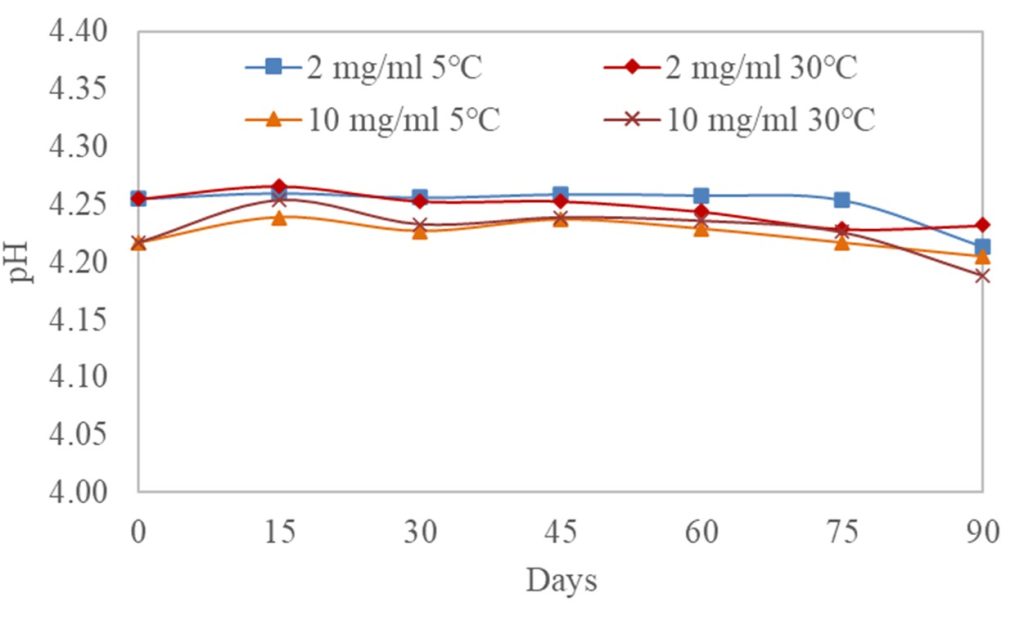
The difference between the results in the 2 mg/mL and 10 mg/mL samples at both storage conditions was statistically proven to be insignificant, with a p-value of 0.856 (p > 0.05) and an F-value of 0.257. The current findings were in agreement with the studies conducted by Svirskis et al. (2020) [9] and Wahab et al. (2020) [13]. In those studies, the physical and chemical stability of furosemide in the commercial vehicle Ora-Blend® was examined. The pH of the suspending system ranged from 4.2 to 7.7, and the formulations were prepared at a concentration of 2 mg/mL. The studies found that extemporaneous formulations of furosemide in Ora-Blend® products were physically and chemically stable over 30 days at 4°C and 25°C. Ora-Blend® SF has an almost identical formulation to X-Temp® Oral Suspension, with a pH buffered to an acidic range of 4.0 – 4.5. Consequently, the results from both studies were unsurprisingly similar. The pH of the furosemide in X-Temp® suspension remained relatively stable throughout the 90-day study, as depicted in Figure II. Both formulations exhibited a consistent pH with a minimal decrease of 0.01 – 0.04 pH units observed from the initial measurement to 90 days.
Building upon the findings of Wahab et al. (2020), which demonstrated that X-Temp® could serve as an appropriate vehicle for furosemide compounded suspension, the current study extended the stability assessment to 90 days. In addition to monitoring the assay percentage of furosemide, the study included a limit test for 4-chloro-5-sulfamoylanthranilic acid, a significant degradation product of furosemide. The content of furosemide and its related compound B were determined quantitatively in this study design. The results indicate that furosemide remains stable in X-Temp® under both refrigerated and real-time storage conditions for the duration of the 90-day study period.
The X-Temp® Oral Suspension System utilise a buffering system consisting of Citric Acid and Monosodium Phosphate. The pH value of furosemide in X-Temp® suspension was stable and remained at around 4.2. According to Kovar et al. [10], furosemide has been observed to undergo acid-catalysed hydrolysis in acidic environments, leading to the breakdown into 4-chloro-5-sulfamoylanthranilic acid and furfuryl alcohol. This instability does not occur in basic conditions, which is essential for the stability of furosemide [4]. Interestingly, despite the general instability in acidic media, stability studies of furosemide in X-Temp® suspension for both concentrations (2 mg/mL and 10 mg/mL) in the present study had shown that even in a low pH system, furosemide remained stable over a period of 90 days. Stability was shown through the limit test (Figure III), which demonstrated that the concentration of the degradation product, 4-chloro-5-sulfamoylanthranilic acid, remained significantly low and well within the acceptable specification limit of 1.5% [7]. Theoretically, changes in pH are often associated with drug degradation [11]. Due to the absence of any considerable pH changes in the extemporaneous preparations in the current study, it could be deduced that no drug degradation had occurred in the formulation.
Furosemide is also known for its photosensitivity, which poses a challenge for its storage and stability [5]. When exposed to light, an aqueous solution of furosemide can undergo photo-induced hydrolysis, consequently resulting in the formation of a yellow solution consisting of the degradation products 4-chloro-5-sulfamoylanthranilic acid and furfuryl alcohol [12]. This degradation not only affects the appearance but can also compromise the efficacy of the medication, which is critical for patients relying on its therapeutic benefits. The use of amber-coloured HDPE bottles in the present study provided a protective barrier against light, effectively shielding the furosemide in X-Temp® suspension from light sources that would otherwise trigger degradation. By safeguarding the medication from light-induced degradation, these bottles ensured that furosemide retained its efficacy throughout the study period, whether stored under refrigerated conditions or at room temperature.
The present research suggests that a furosemide oral suspension utilising the X-Temp® Oral Suspension System to suspend furosemide offers a promising treatment alternative for patients, particularly children, who have difficulty swallowing tablets.
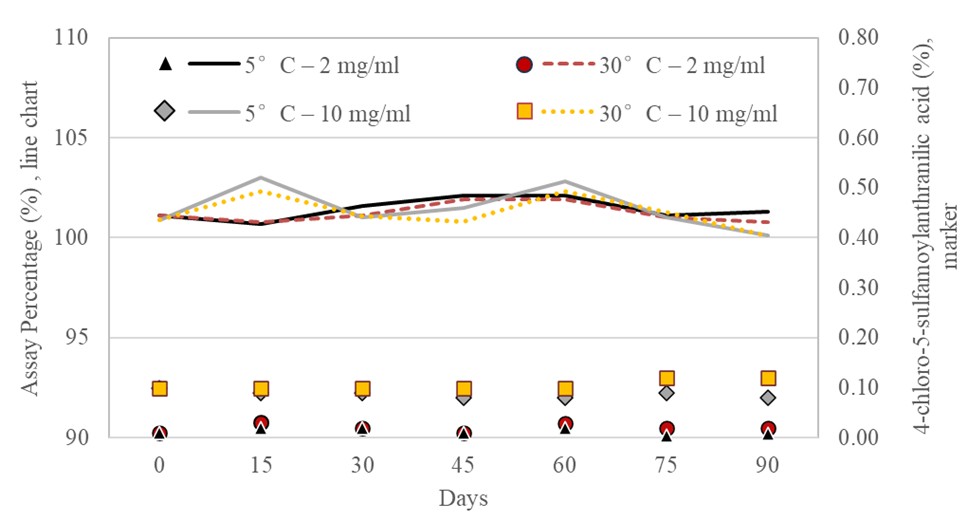
The study finds that an oral suspension with a concentration as low as 2 mg/mL and as high as 10 mg/mL can remain stable for up to 90 days, regardless of whether it was kept at room temperature or refrigerated, potentially enhancing the accessibility of the medication and ease of administration.
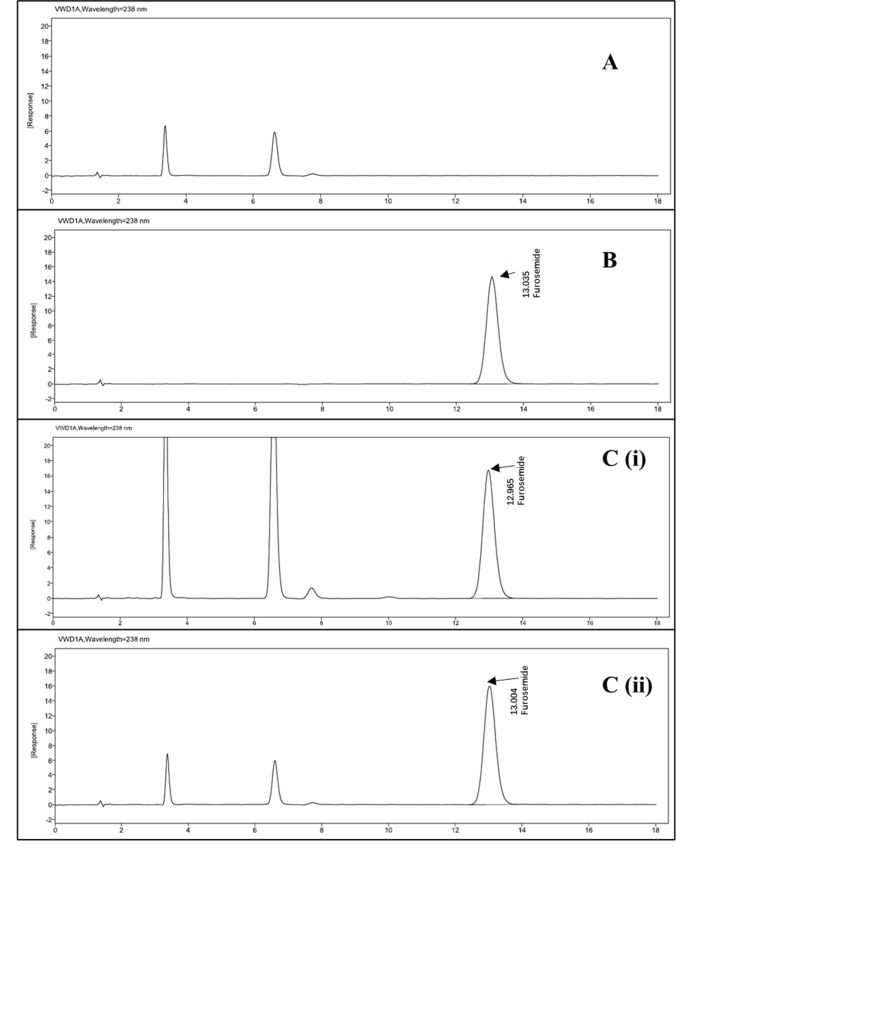
The chromatograms (Figure IV) illustrated the selectivity of the HPLC method utilised in this research, showing insignificant interference from the excipients present in the formulation. The chromatograms of the analysed samples at various time points during the stability study showed that there was no additional peak correlated to any potential degradation products.
Microbiological Stability
Based on the results in Table 4 and 5, none of the study samples exhibited any microbial growth throughout the entire study duration. The microbial analysis indicated that TAMC, TYMC, and E. coli were not detected, which evinced the adherence to the established quality requirements. This was indicative of the potential of the X-Temp® Oral Suspension System in maintaining the microbial stability of the suspension over the course of the study. The suspensions were maintained at around pH 4.2, which contributed to the effectiveness of the preservatives and provided an environment that hindered microbial growth. This aspect is of pivotal importance since the shelf-life of many extemporaneous formulations is often restricted by the susceptibility to microbial growth, on top of the stability of the active component itself.
CONCLUSION
Furosemide oral suspensions consisting of 2 mg/mL and 10 mg/mL of furosemide were readily formulated from commercially available tablets using the X-Temp® Oral Suspension System. The compounding exhibited reassuring stability for up to 90 days while preserving all the quality characteristics. Based on the results, it can be concluded that furosemide in X-Temp® suspension at concentrations ranging from 2 mg/mL to 10 mg/mL demonstrates physical, chemical, and microbiological stability under refrigeration and at room temperature for a minimum of 90 days when stored in amber HDPE bottles. This formulation, prepared using the X-Temp® Oral Suspension System, offers a promising alternative in terms of efficacy, safety, and reliability for hospital pharmacy services to optimise paediatric diuretic therapy.
The stability study results indicated that the X-Temp® Oral Suspension System could serve as a stable suspending vehicle for compounding various active pharmaceutical ingredients indicated for a wide range of medical applications. In addition to providing a stable formulation, it offers several other benefits, such as being alcohol-free, colorant-free, and sugar-free.
ACKNOWLEDGEMENT
The authors acknowledge the contribution of Pharm-D Health Science Sdn. Bhd. for supplying the X-Temp® Oral Suspension System, furosemide tablets and plastic HDPE bottles used in this study.
CONFLICT OF INTEREST
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
REFERENCE
- Batchelor, H.K. & Marriott, J.F. Formulations for children: Problems and solutions. Br J Clin Pharmacol, 2015; 79(3): 405-418. https://doi.org/10.1111/bcp.12268
- Kearns, G.L., Abdel-Rahman, S.M., Alander, S.W., Blowey, D.L., Leeder, J.S., & Kauffman, R.E. Developmental pharmacology—Drug disposition, action, and therapy in infants and children. The New England Journal of Medicine, 2003; 349(12):1157-1167. https://www.nejm.org/doi/full/10.1056/NEJMra035092
- Costello, I., Long, P.F., Wong, I.K., Tuleu, C. & Yeung, V. Paediatric drug handling. Pharmaceutical Press, London. 2007 https://journals.sagepub.com/doi/10.1345/aph.1K108
- Ghanekar, A.G., Gupta, V.D. & Gibbs, C.W. Stability of furosemide in aqueous systems. J Pharm Sci, 1978; 67(6):808-810. https://doi.org/10.1002/jps.2600670621
- Moore, D.E. & Sithipitaks, V. Photolytic degradation of frusemide. J. Pharm. Pharmacol, 1983;35(8): 489-493. https://doi.org/10.1111/j.2042-7158.1983.tb04816.x
- British Pharmacopoeia Commission. British Pharmacopoeia 2024 Volume V. Appendix XVIB. London, The Stationery Office. 2023 http://dx.doi.org/10.52494/maljpharmv10208
- USP. Furosemide Oral Solution. In: USP-NF. 2022 [cited May 1, 2022]. Available from https://doi.org/10.31003/USPNF_M34560_03_01
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonised Tripartite Guideline, Q2 (R2). [cited May 1, 2024]. Available from https://database.ich.org/sites/default/files/ICH_Q2-R2_Document_Step2_Guideline_2022_0324.pdf
- Svirskis, D., Jaffer, J., Agarwa, P., Khan, A., Kaur, J., Cheng, A. & Hanning, S. Alcohol-free extemporaneous formulations of furosemide are chemically and physically stable in Ora-Blend products for 30 days. Int J Pharm Compd, 2020;24(3):246-251. https://pubmed.ncbi.nlm.nih.gov/32401745
- Kovar, K.A, Wojtovicz, G.P. & Auterhoff, H. Hydrolysis of diuretic sulphonamides. Archiv der Pharmazie, 1974; 307(74): 657-662. https://doi.org/10.1002/ardp.19743070902
- Shoosanglertwijit, J., Kaewnopparat, S., Yongmaitreesakul, B., Pattayananthavej, S., & Kaewnopparat, N. Physical, chemical, and microbiological stability of extemporaneous furosemide suspensions. Asian Biomed, 2011; 5(5):681-686. https://intapi.sciendo.com/pdf/10.5372/1905-7415.0505.089
- Giannetti, M., Canale, V.C., Micheli, L., Fiori, M., Mazzuca, C. & Palleschi, A. An insight into the degradation processes of the anti-hypertensive drug furosemide. Molecules, 2023; 28(1):381. https://doi.org/10.3390/molecules28010381
- Wahab, M. F. B. A., Amir, A. B., Razi, S. S. B. M., & Ramli, N. B. The Stability Study of Extemporaneous Preparations Prepared in The Outpatient Pharmacy of Tuanku Fauziah Hospital Stored in Patient’s Setting. Pharm. Res Reports, 2020; 40. https://research.pharmacy.gov.my/sites/default/files/publication/PRR%20Vol3%202020%20Issue2.pdf#page=46
Please cite this article as:
Freeda Siew Yuin Thean, Lucy Yeoh, Rou Chian Ng and Ke X. Thong, Stability study of an extemporaneous furosemide oral suspension prepared using commercially available tablets with X-Temp® Oral Suspension System. Malaysian Journal of Pharmacy (MJP). 2024;2(10):56-65. https://mjpharm.org/stability-study-of-an-extemporaneous-furosemide-oral-suspension-prepared-using-commercially-available-tablets-with-x-temp-oral-suspension-system/